Mayer-Rokitansky-Küster-Hauser Syndrome: Rare case diagnosis and management
Rokade J1, Upadhye Gaurav P2*, Shrivastav K3, Zofi M4
DOI:https://doi.org/10.17511/joog.2025.i01.02
1 Jyoti Rokade, Associate Professor, Department of Obstetrics and Gynecology, Government Medical College Miraj, Maharashtra, India.
2* Prachi Gaurav Upadhye, MCh Plastic and Reconstructive Surgeon, Assistant Professor, Department of General Surgery, Government Medical College Miraj, Maharashtra, India.
3 Kiran Shrivastav, Assistant Professor, Department of Obstetrics and Gynecology, Government Medical College Miraj, Maharashtra, India.
4 Misbah Zofi, Junior Resident, Department of Obstetrics and Gynecology, Government Medical College Miraj, Maharashtra, India.
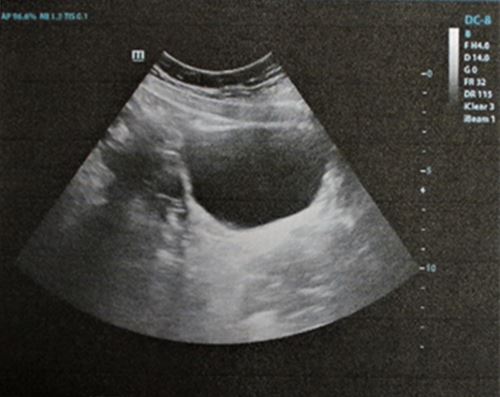
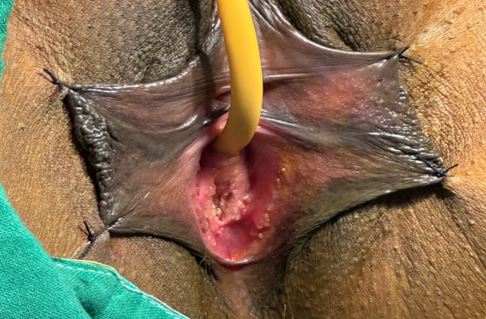
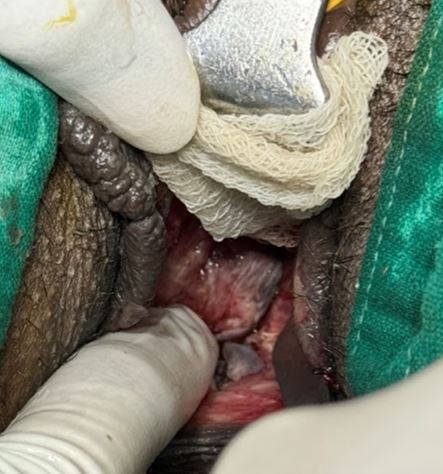
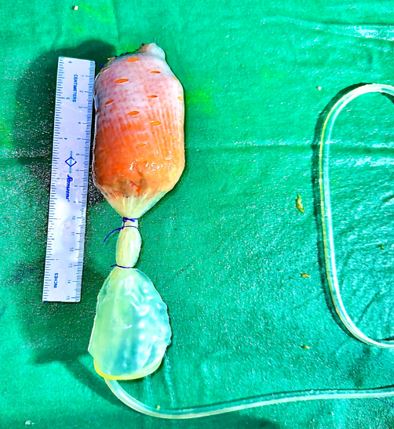
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a rare congenital disorder characterized by vaginal agenesis or hypoplasia, often associated with uterine anomalies. We present a case of a 35-year-old woman with Type 1 MRKH syndrome, who underwent McIndoe vaginoplasty to create a functional vagina. The patient presented with primary amenorrhea and difficulty with sexual intercourse. Preoperative evaluation confirmed vaginal agenesis, and the patient underwent McIndoe vaginoplasty using a split-thickness skin graft. Postoperative follow-up showed successful creation of a functional vagina, with satisfactory anatomical and functional outcomes. This case highlights the effectiveness of McIndoe vaginoplasty in creating a functional vagina in patients with MRKH syndrome, improving their quality of life and sexual function.
Keywords: MRKH syndrome, vaginal agenesis, McIndoe vaginoplasty, vaginoplasty, congenital disorder
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , MCh Plastic and Reconstructive Surgeon, Assistant Professor, Department of General Surgery, Government Medical College Miraj, Maharashtra, , India. Email:  |
Rokade J, Upadhye Gaurav P, Shrivastav K, Zofi M, Mayer-Rokitansky-Küster-Hauser Syndrome: Rare case diagnosis and management. Obs Gyne Review J Obstet Gynecol. 2025;11(1):6-9. Available From https://obstetrics.medresearch.in/index.php/joog/article/view/179 |
 |
 ©
©